【亚马逊|机器学习解决了谁是谁的问题】
固体核磁共振(NMR)光谱--一种测量在强磁场中暴露于无线电波的一些原子核所发出的频率的技术 , 可用于确定化学和三维结构以及分子和材料的动力学 。
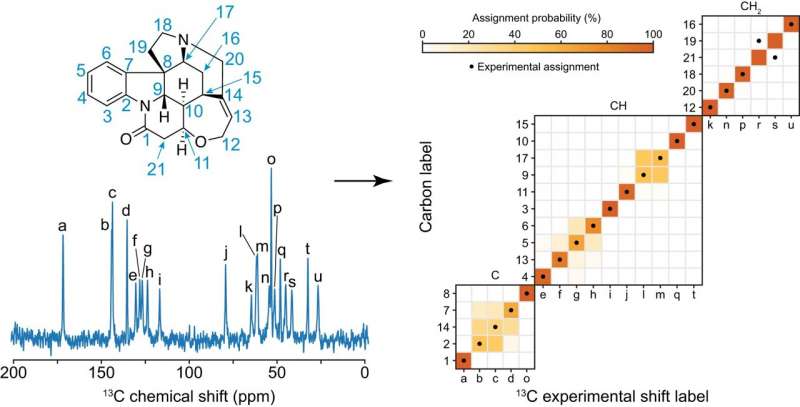
分析中一个必要的初始步骤是所谓的化学位移分配 。 这涉及到将核磁共振频谱中的每个峰值分配给正在调查的分子或材料中的一个特定原子 。 这可能是一项特别复杂的任务 。 以实验方式分配化学位移可能具有挑战性 , 通常需要进行耗时的多维相关实验 。 通过对比实验化学位移数据库的统计分析进行分配将是一个可供选择的解决方案 , 但目前还没有针对分子固体的这种数据库 。
一个研究小组 , 包括EPFL教授Lyndon Emsley(磁共振实验室负责人)、Michele Ceriotti(计算科学和建模实验室负责人)和博士生Manuel Cordova , 决定通过开发一种直接从二维化学结构中概率地分配有机晶体的核磁共振光谱的方法来解决这个问题 。
他们首先创建了自己的有机固体化学位移数据库 , 将剑桥结构数据库(CSD)与ShiftML相结合 , 后者是一个包含20多万个三维有机结构的数据库 , 是他们之前共同开发的机器学习算法 , 可以直接从分子固体的结构中预测化学位移 。
最初在2018年的《自然-通讯》论文中描述 , ShiftML使用DFT计算进行训练 , 但随后可以对新结构进行准确的预测 , 而无需进行额外的量子计算 。 虽然达到了DFT精度 , 但该方法可以在几秒钟内计算出约100个原子的结构的化学位移 , 与目前的DFT化学位移计算相比 , 计算成本降低了1万倍之多 。 该方法的准确性并不取决于所研究结构的大小 , 预测时间与原子数呈线性关系 。 这为在以前不可行的情况下计算化学位移创造了条件 。
在新的《科学进展》论文中 , 该团队使用ShiftML预测了从CSD中提取的20多万个化合物的位移 , 然后将获得的位移与分子环境的拓扑表示联系起来 。 这涉及到构建一个代表分子中原子之间共价键的图形 , 将其延伸到远离中心原子的特定数量的键 。 然后 , 他们把数据库中所有相同的图的实例集中起来 , 使他们能够获得每个图案的化学位移的统计分布 。 该表示法是对分子中原子周围的共价键的简化 , 不包含任何三维结构特征:这使他们能够通过结合分子中所有原子的分布的边际化方案 , 直接从有机晶体的二维化学结构中获得其核磁共振谱的概率分配 。
在构建了化学位移数据库之后 , 科学家们着眼于预测模型系统的分配 , 并将该方法应用于一组有机分子 , 这些分子的碳化学位移分配至少有一部分已经通过实验确定:茶碱、百里酚、可卡因、士的宁、AZD5718、利西诺普利、利托那韦和青霉素G的K盐 。
最后 , 他们在100个具有10到20个不同碳原子的晶体结构的基准上评估了该框架的性能 。 他们使用ShiftML预测的每个原子的位移作为正确的分配 , 并把它们从用于分配分子的统计分布中排除 。 在超过80%的情况下 , 在两个最可能的分配中发现了正确的分配 。
\"科尔多瓦说:\"这种方法可以通过简化这些研究的重要第一步 , 大大加快核磁共振的材料研究 。
- 亚马孙热带雨林|“地球之肺”亚马逊雨林,为什么是人类禁区?到底有多恐怖?
- 亚马逊|2019年,一头座头鲸出现在亚马逊雨林中,生物学家百思不得其解!
- 机器|激光粉末床熔合中成分和相图特征对适印性和微观结构的影响:合金系统加工图的开发和比较 (一)
- 机器|也许未来,人类进化的方向是成为机器人,人族进化成机器族
- 机器人|得物App公布“潮流主场计划”:3.2亿现金,200亿流量扶持潮流内容创作者
- 机器人|?我国拿下全球首例!售价2000万机器立大功,前沿技术冲至领先
- 超星系团|3亿公里外,仅能工作17小时的德国机器人,传回令人欣喜的照片
- 华为|华为平板电脑,办公学习都用它!
- 机器|儿童用药:实验室ICP-MS检测,药物中是否含重金属元素
- 郑州|黑科技!郑州各汽车站上线智能测温机器人